



MED-SuMo software
The Protein Data Bank PDB is a unique public source of macromolecular structures with co-crystallized ligand(s) in many cases. MED-SuMo software makes possible to compare and superpose any 3D interaction molecular surface accross the PDB. It opens new approaches in structure based drug design to molecular modelers, crystallographers and medicinal chemists. MED-SuMo retrieves onto your binding binding site of interest or a defined protein full surface, all PDB structures that are sharing in 3D a network of intermolecular interactions such as charges, H-bonds, hydrophobic and aromatic stacking.
Heuristic:
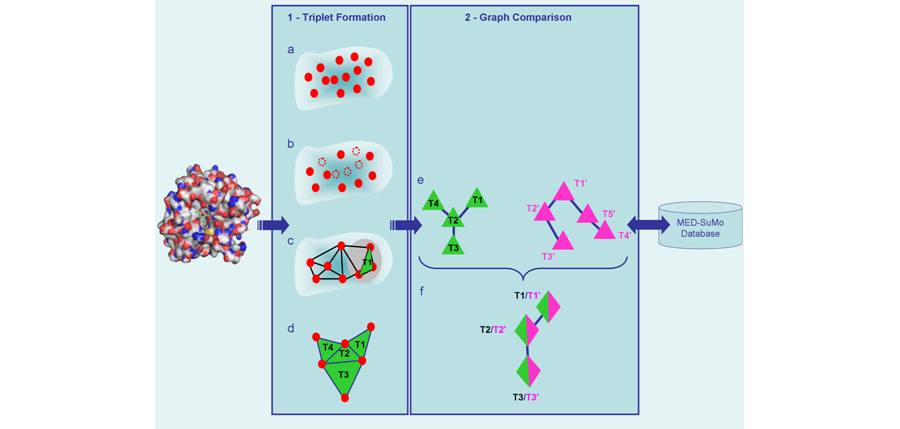
Surface chemical features: in MED-SuMo, the notion of Chemical Features is fundamental. Every macromolecular structure is first converted into a set of chemical features. Only Features which are available for interacting with ligands are selected and are named Surface Chemical Features (SCF). Several types of chemical features are defined by default but can be easily modified. Each of them will represent a given property. Only chemical features of the same type can be compared and possibly considered as equivalent. The default dictionary of SCFs such as H-Bonds, formal charges, hydrophobic and aromatic groups is shown below in Fig.1.
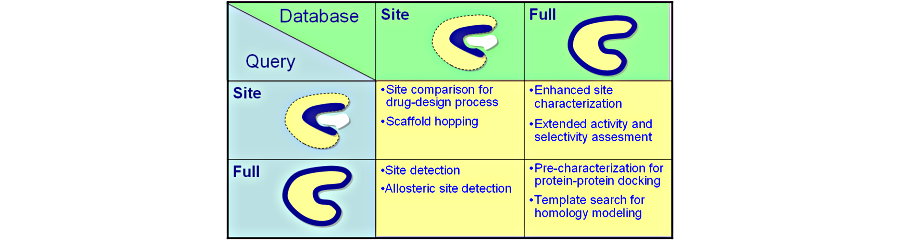
Two kinds of MED-SuMo databases: sites database and full surface databases: in Drug Design applications, it’s the best technology to take advantage of the ongoing exponential growth of the public Protein Data Bank[3] where all 3D experimental protein-ligands are stored. MED-SuMo is a fast and reliable technology to query and mine the biggest available macromolecules 3D structural database.
The reference database, the Protein Data Bank is freely and publicly available and contains (update april 2008) the 3D atomic coordinates of:
– 136,000 ligands bound to macromolecules (8,000 are distinct)
– 50,000 macromolecules (47,000 proteins)
A MED-SuMo site database contains the graphs of SCFs triplets which are in the environnement of a ligand. This environnement is defined by a maximum distance between atoms of the ligand and chemical features of 4.5 Å or 6.0 Å in most cases (user defined). 6.0Å corresponds to a broader binding site definition around a ligand and is a better choice for site detection and functionnal annotation. 4.5 Å is more suited to drug design applications. A ligand is defined as a set of heteroatoms or small peptides. A full surface database corresponds to the whole surface, e.g. all features but burried or involved in intraprotein h-bonds. The corresponding MED-SuMo databases encodes the whole PDB (as april 2008):
– Site database containing 136,000 ligand sites description
– Full surface database which contains 50,000 full surface description (protein, RNA and DNA can be described)
4 comparison modes can be exploited:
MED-SuMo server parameters: the triangle network of chemical features can be tuned to be more or less dense. The maximum length of an edge and the maximum sum of the three edges are tunable parameters when the database is generated:
– High density triangle network (BEST): parameters 20-60
– Default density triangle network (FAST): parameters 13-39
When surface chemical matches, they are eventually tested for having a similar shape environment. This shape threshold is a tunable parameter when the comparison is ran:
– Default shape threshold: ST=65%
– Lower shape threshold to allow more tolerance in the shape comparison: ST=45%
References:
[1] Jambon M, Imberty A, Deléage G, Geourjon C “A new bioinformatic approach to detect common 3D sites in protein structures” Proteins: Struct., Funct., and Gen. 52:137-145 (2003)
[2] Jambon M, Andrieu O, Combet C, Deléage G, Delfaud F, Geourjon C “The SuMo server : 3D search for protein functional sites” Bioinformatics Vol 21, n°20, 3929-3930 (2005)
[3] H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, P.E. Bourne: “The Protein Data Bank” (2000) Nucleic Acids Research, 28 pp. 235-242.
Working with MED-SuMo:
Input: MED-SuMo uses as input a macromolecular structure database with up to hundreds of thousands structures:
– the PDB (default)
– in house curated database
– models
Output:
– Ligand are aligned, shown in the graphical user interface and output as they are defined in the original PDB file.
– In house curation of PDB ligands can be applied on the output of MED-SuMo.
– MEDIT is providing by default an optional bond order and aromatic perception for PDB ligands.
– Superposed ligand and/or proteins can be exported in PDB, SDF and Mol2 file format.
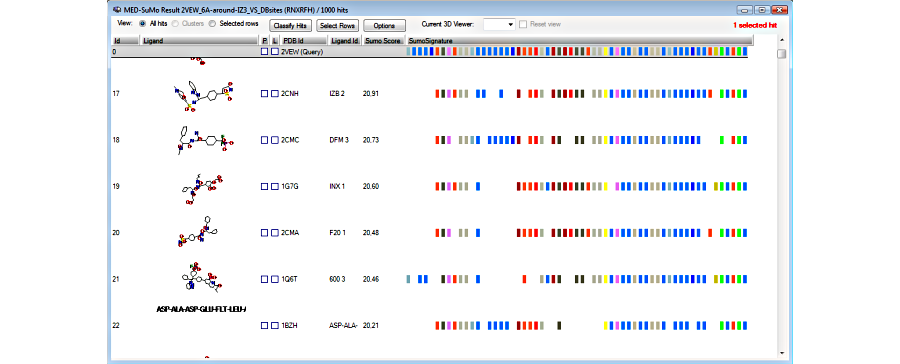
Find the most interesting hits with the MED-SuMo score:
Working with MED-SuMo is easy and fast. Simply by clicking, superpositions of structures and fragments can be displayed in the 3D viewer. Hits are retreived and ranked in a spreadsheet according to the MED-SuMo score which takes into account chemical features and shape and is very efficient to identify the best hits:
– Only true positives are found on the top of list (MED-SuMo score > 6.0)
– Interfamily hits are found from MED-SuMo score 6.0 to 3.0
– False positives have a drastically different signature than true positives and they can easily be filtered out
MED-SuMo server:
– Runs on linux systems
– Takes advantage of the most recent hardware architectures: multicore processors and computer clusters
– Command Line User Interface (CLUI) with LUA scripts
– MED-SuMo databases are easy to update from the PDB website
MED-SuMo graphical user interface GUI:
– Runs on Windows: XP, Vista, Seven, 8, 10
The GUI features:
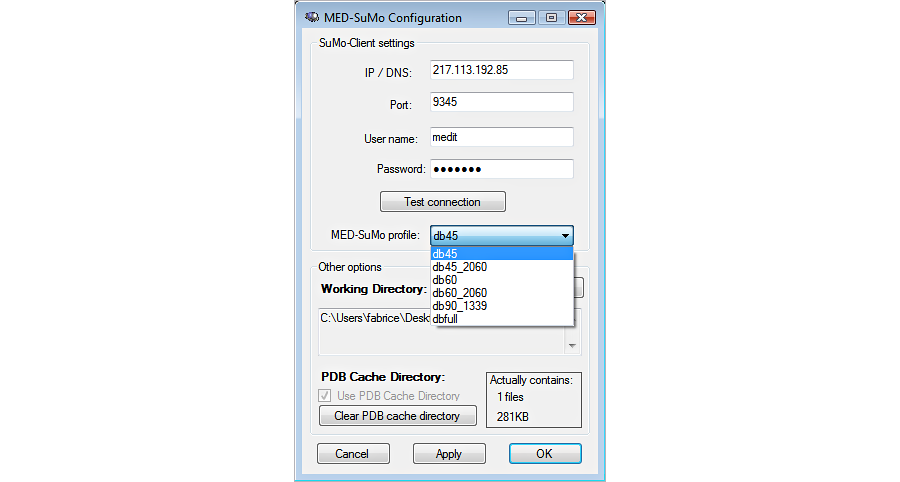
1. Connect to the server
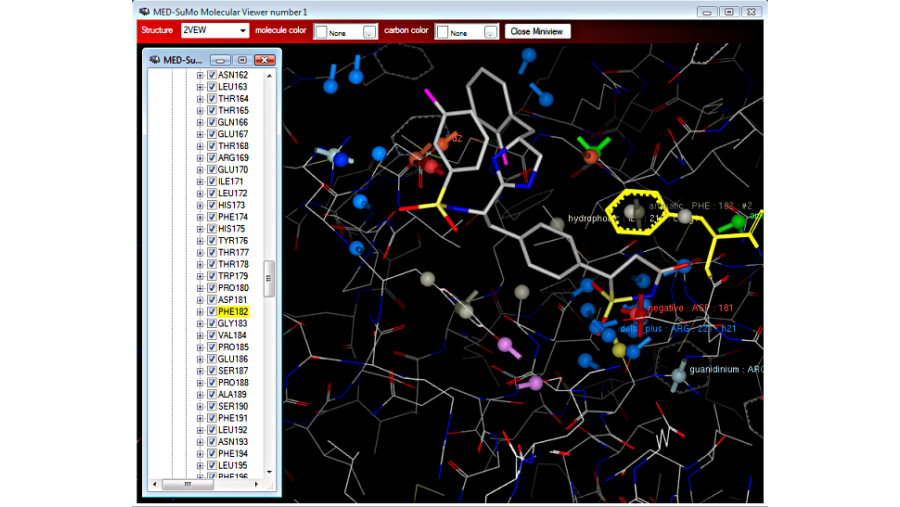
2. Build the query : The query is a surface of a protein (locally stored or from the database). The surface is described by a graph of triplets of surface chemical features.
Visualize the triplets of surface chemical features:
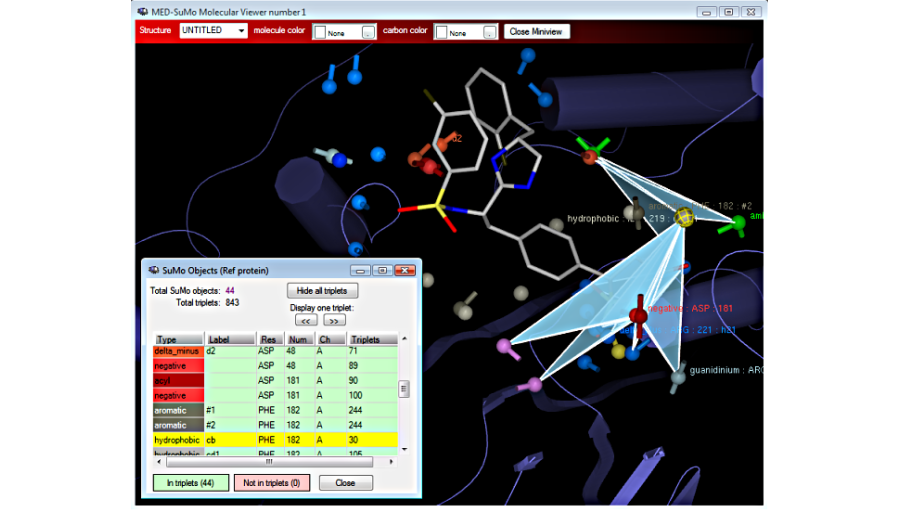
Choose residues or atoms to define the query or change the rendering
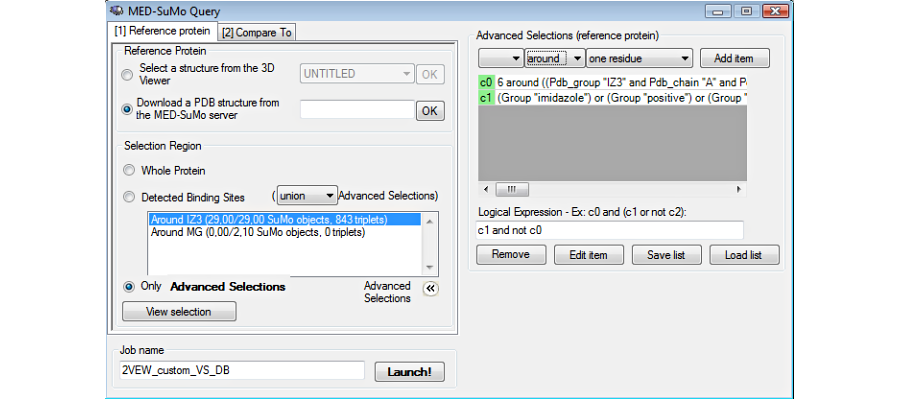
Choose the query with auto-detected sites:
Choose the query by defining an user advanced selection:
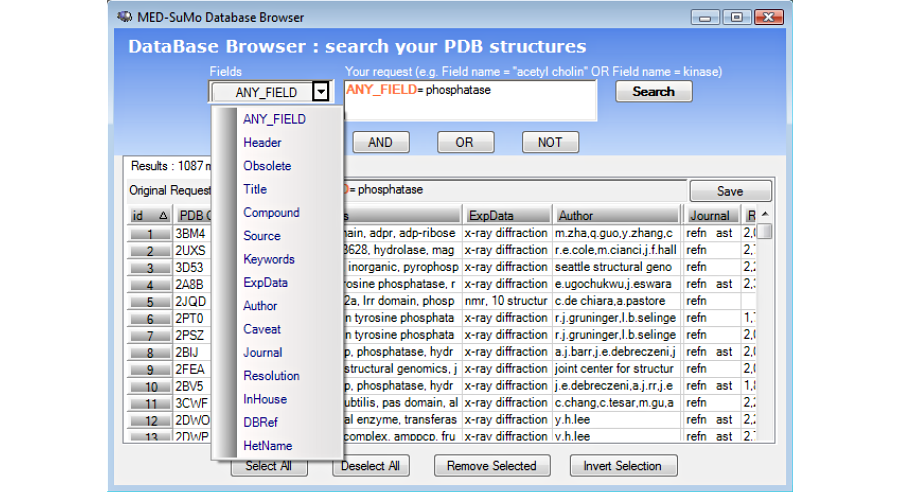
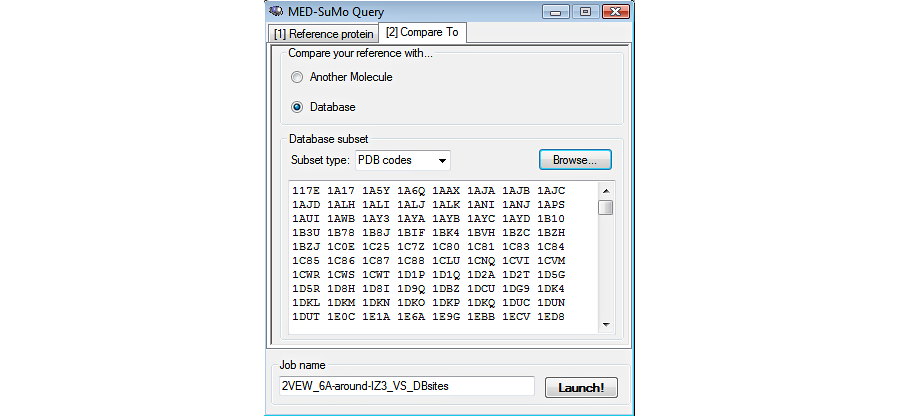
Choose the database: The MED-SuMo databases (Sites or Full) are precomputed. The user chooses the database at the moment of the connection. Then chooses the structures of the database: whole database, subset only, a single locally stored structureBefore launching the job, the user can select a subset of the database or compare the query to a single file stored locally).
3. Browse the results in the result table
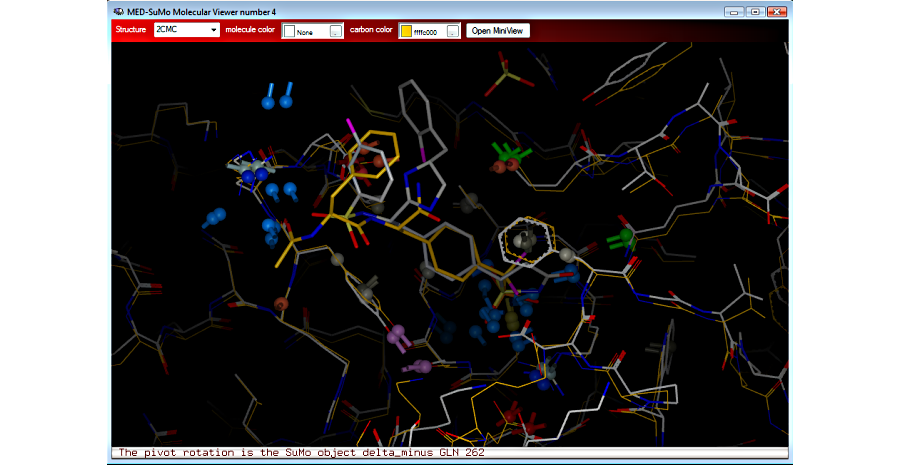
4. View hits superposed to the query in the 3D viewer
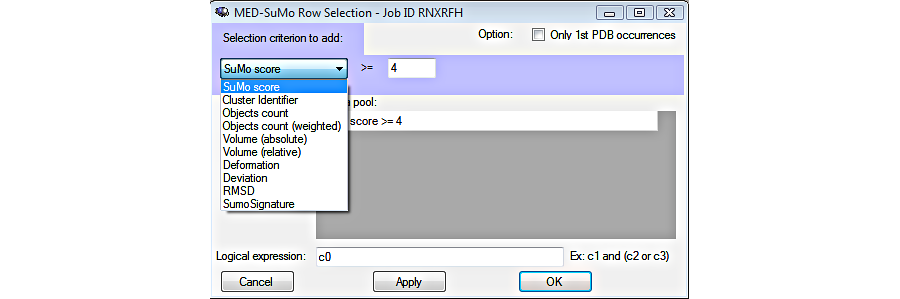
5. Sort and select results
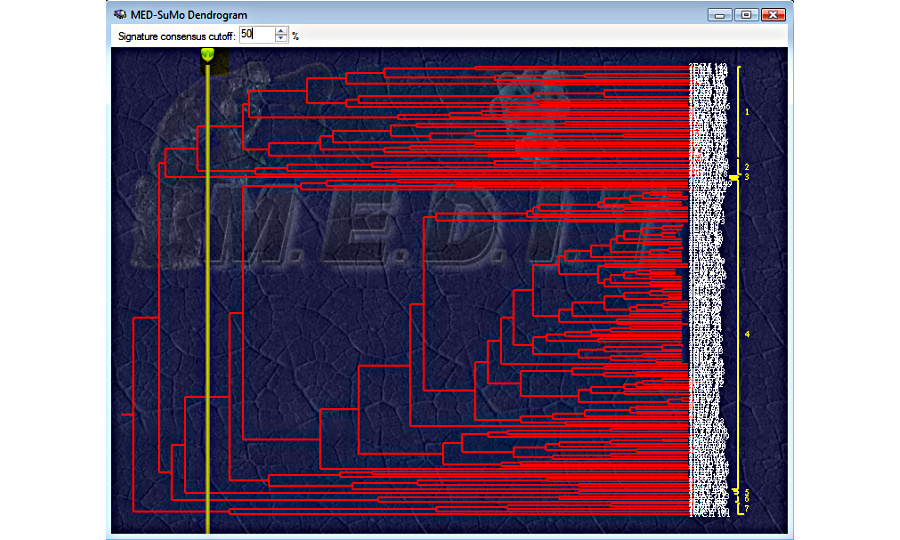
6. Selected hits can be clustered according to their SCF signature
MEDP-Fragmentor:
MEDP-Fragmentor is part of the MED-SuMo fragment-based technology and performes the generation of the MED-SuMo fragment database from a database like the PDB.
The MED-portions are encoded in the MED-SuMo fragment database which is ready to be searched and browsed with MED-SuMo. These 3D patterns, called MED-Portions, include chemical moieties which are matching molecules from a chemical library and substructures of protein-bound ligand. MED-Portions, which are the MED-SuMo representation of protein-fragment patterns defined by several criteria: (1) a chemical moiety where atoms are topologically matching with a molecule from molecular libraries, e.g. synthetically accessible molecules or building blocks, (2) open valences filled by ‘dummy atoms’ that indicate where it was connected in the original ligand, and (3) the protein interaction surface surrounding that chemical moiety described by the MED-SuMo Surface Chemical Features (SCFs).